Introduction
Monoclonal antibodies (mAbs) have become the biggest and fastest-growing class of biotherapeutics, addressing diverse indications ranging from oncology and immunology to infectious and rare diseases (1–3). With increasing patient populations and a shift toward long-term self-administered therapies, subcutaneous (SC) delivery of mAbs is increasingly preferred over intravenous (IV) infusion. Compared with IV dosing, SC administration reduces treatment burden, improves patient convenience, and enables health-system efficiencies by minimizing infusion-center visits (4, 5). However, delivering therapeutic doses of mAbs within the limited SC injection volume (usually ≤2 mL and up to 5–10 mL with special technologies) requires the development of high-concentration formulations, often above 100–150 mg/mL (6). Formulating mAbs at concentrations exceeding 100–150 mg/mL for SC delivery introduces a unique collection of biophysical, chemical, process, and regulatory challenges not observed at lower doses. Strong protein–protein interactions can increase viscosity. They can also cause protein self-association, opalescence, and liquid–liquid phase separation (LLPS). These effects can make manufacturing difficult. They can also reduce syringeability and compatibility with prefilled syringes, autoinjectors, and wearable delivery devices (7–9). These problems can become worse due to chemical degradation such as deamidation, oxidation, and glycation. Physical instability like aggregation, particle formation, and protein unfolding can also occur. Together, these issues can reduce drug potency, increase product variability, and raise the risk of immunogenicity (10, 11). While conventional excipients (e.g., sugars, surfactants, and amino acids) provide partial mitigation, their protective capacity diminishes at extreme concentrations, and the regulatory pathway for novel stabilizers remains restrictive. From a processing perspective, ultrafiltration and diafiltration can be affected by membrane fouling and shear sensitivity. Filling operations become difficult because high-viscosity solutions flow slowly. Container closure systems may also cause problems such as protein adsorption, leachables, and interactions with silicone oil. In addition, device and patient factors must be considered, such as injection volume, injection force, and ease of use. Together, these challenges make high-concentration mAb formulation complex. Therefore, an integrated approach is needed, including protein engineering, careful excipient selection, optimized process design, predictive modeling, and risk-based control strategies to ensure product quality, stability, and usability (11).
To overcome challenges associated with high-concentration monoclonal antibody formulations, several strategies focus on controlling the molecular factors responsible for viscosity and instability. Amino-acid excipients such as arginine and histidine can reduce attractive protein–protein interactions, while adjustment of pH and ionic strength helps regulate electrostatic forces and minimize aggregation. Protein engineering can be used to modify surface charge or reduce hydrophobic regions of antibodies. This helps improve antibody developability and reduce viscosity. In addition, the relationship between protein concentration and solution rheology helps scientists better understand these systems. As antibody concentration increases, viscosity typically rises in a nonlinear manner due to enhanced intermolecular interactions, which can be described using models such as the Krieger–Dougherty or Mooney equations. In addition, parameters such as the second virial coefficient (B22) and the diffusion interaction parameter (kD), obtained from light-scattering techniques, provide quantitative indicators of protein–protein interactions and aggregation propensity (12, 13).
Differences among antibody subclasses (e.g., IgG1, IgG2, and IgG4) may also influence rheological behavior because of variations in structural flexibility, disulfide bonding patterns, and surface charge distribution. Furthermore, solvent composition and excipient selection can significantly modulate intermolecular interactions by affecting electrostatic screening, hydration layers, and protein stability. Collectively, these quantitative and molecular insights support a more rational and predictive approach to the design of stable, low-viscosity, high-concentration monoclonal antibody formulations (14, 15).
Recent advances have significantly expanded the mechanistic understanding of the molecular drivers of viscosity and instability, including antibody sequence and structural motifs, surface charge distribution, glycosylation, and excipient interactions (16–18). In parallel, predictive approaches—ranging from empirical correlations to artificial intelligence (AI) and machine learning (ML) models that integrate multi-modal datasets—are increasingly being used to anticipate high-concentration behavior earlier in development (19–21). Complementary strategies, including protein engineering, excipient innovation, process optimization, and the use of recombinant human hyaluronidase PH20 (rHuPH20) to enable large-volume subcutaneous (LVSC) administration, are also being explored to mitigate risks and expand dosing feasibility (22–24).
This review summarizes the current understanding of high-concentration monoclonal antibody (mAb) formulation science, with emphasis on the mechanistic basis of viscosity and instability, emerging predictive analytics, formulation and process engineering strategies, delivery device considerations, and regulatory expectations within Quality by Design (QbD) and risk-based control frameworks (25, 26). Through selected case studies and practical development insights, the review highlights rational approaches to mitigate developability risks associated with concentrated antibody solutions. Key areas discussed include the molecular determinants governing high-concentration rheology, experimental and computational tools for predicting viscosity and protein–protein interactions, and formulation strategies—such as excipient optimization and protein engineering—to reduce viscosity while maintaining structural stability. The review also addresses container–closure interactions, device compatibility, and regulatory considerations necessary for establishing robust control strategies that ensure product quality and patient usability. Finally, emerging technologies and translational opportunities are discussed, outlining future directions for the development of next-generation, patient-centric antibody therapeutics.
Molecular determinants of viscosity at high protein concentration
The rheological behavior of mAbs at high concentrations is mainly governed by short- and long-range intermolecular interactions that promote solution structuring and molecular crowding. Unlike dilute solutions, where Brownian motion and solvent interactions dominate, concentrated protein solutions exhibit complex colloidal behavior. As protein concentration increases and protein–protein interaction thresholds are exceeded, viscosity rises sharply in a nonlinear manner (27–29). Table 1 and Figure 1 summarize molecular determinants of viscosity in high-concentration mAb formulations.
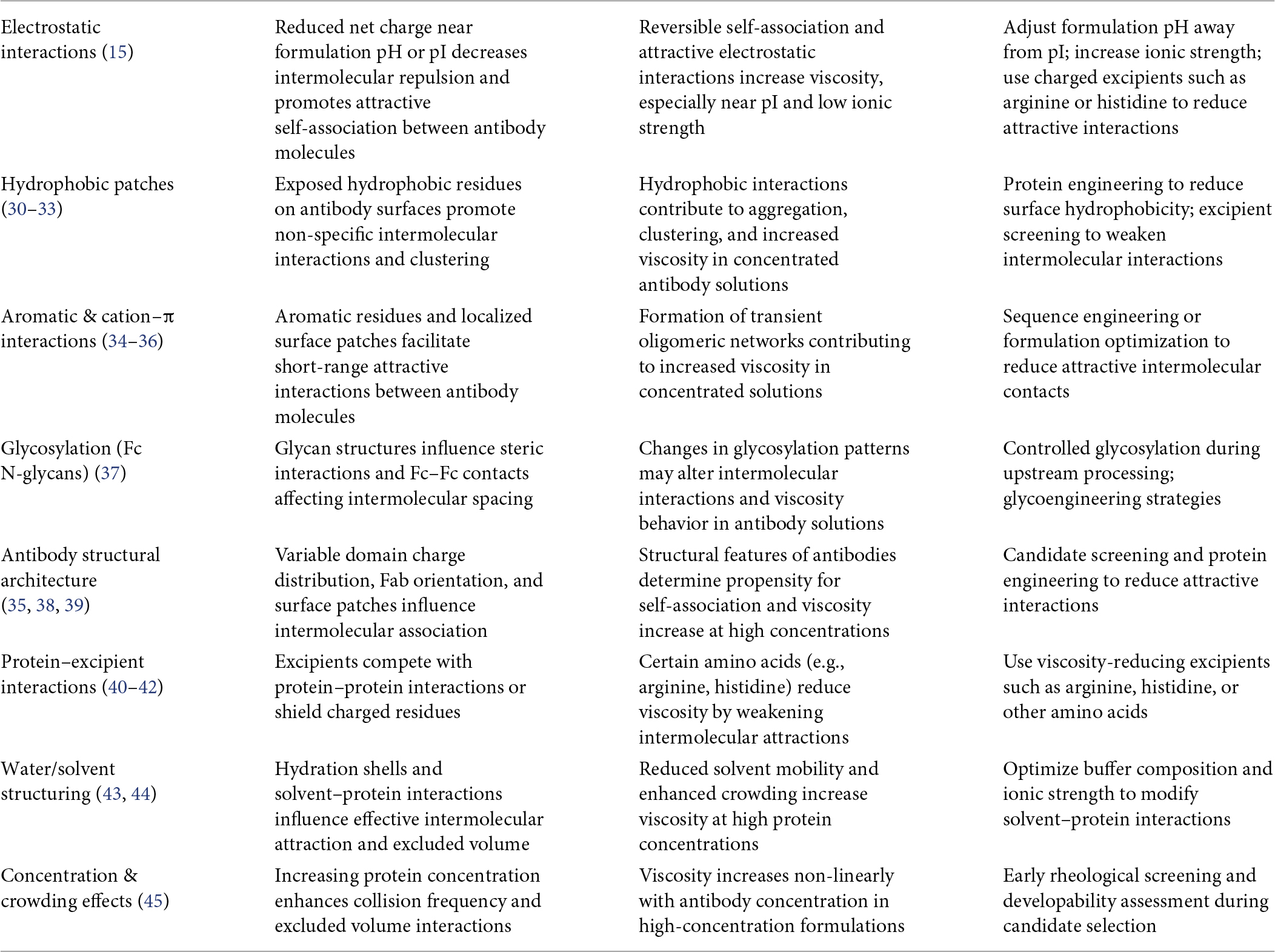
Table 1. Molecular determinants of viscosity in high-concentration mAb formulations.
Figure 1. Molecular determinants of viscosity in high-concentration monoclonal antibody formulations.
Molecular determinants
Electrostatic interactions
• Charge distribution and isoelectric point (pI): Antibodies with pI values close to the formulation pH have lower net charge, which reduces electrostatic repulsion and promotes attractive interactions and clustering.
• Charge patches: Localized positive or negative regions on the antibody surface create heterogeneous interaction fields that promote reversible self-association and network formation.
• Ionic strength effects: Increased salt concentration can screen electrostatic repulsion and accelerate viscosity increase, while optimized ionic conditions may help suppress attractive interactions.
Electrostatic interactions—determined by pI, surface charge distribution, and ionic strength—play an important role in controlling reversible self-association and viscosity behavior in high-concentration mAb formulations (15).
Hydrophobic interactions
• Surface hydrophobic patches: Exposed aromatic or aliphatic residues on antibody surfaces promote intermolecular contacts and non-specific protein association.
• Temperature sensitivity: Hydrophobic interactions become stronger at higher temperatures, which can increase viscosity and promote opalescence under physiologically relevant conditions.
Hydrophobic interactions, mainly arising from exposed surface patches and enhanced by temperature, promote non-specific self-association and contribute significantly to viscosity and opalescence in high-concentration mAb formulations (30–33).
Aromatic and cation–π interactions
• Aromatic residues (Tyr, Phe, and Trp): π–π stacking and cation–π interactions between aromatic side chains in Fc and Fab domains, or between neighboring antibody molecules, can promote reversible oligomerization.
• Concentration dependence: These interactions are relatively weak at low concentrations but become more significant at concentrations above ∼100 mg/mL, where they contribute to transient network formation that limits molecular mobility.
Aromatic and cation–π interactions involving Tyr, Phe, and Trp residues can contribute to reversible oligomerization and transient network formation in concentrated antibody solutions, thereby increasing viscosity and restricting molecular mobility (34–36).
Glycosylation and post-translational modifications
• Fc N-glycans: Glycans attached to the Fc region can influence intermolecular interactions. They may sterically hinder close molecular packing and reduce viscosity or, in some cases, stabilize antibody self-association depending on glycan structure.
• Heterogeneous glycoforms: Variations in glycan structures can alter solution behavior and may contribute to variability in viscosity profiles between production batches.
Fc glycosylation and other post-translational modifications can modulate antibody self-association by either sterically hindering or stabilizing intermolecular contacts. In addition, glycoform heterogeneity may introduce batch-to-batch variability in viscosity and overall solution behavior in concentrated mAb formulations (37).
Antibody structural architecture
• Domain arrangement and flexibility: Antibodies with more open conformations or long, flexible complementarity-determining region (CDR) loops may have a greater tendency for intermolecular self-interactions.
• Fc–Fab orientation: The spatial arrangement and angle between Fc and Fab domains can influence steric hindrance and affect molecular crowding and interaction dynamics.
• Isotype differences: Different IgG subclasses exhibit distinct rheological behavior; for example, IgG1 and IgG4 often show higher viscosity than IgG2 at comparable concentrations due to differences in structural flexibility and intermolecular interactions.
Antibody structural architecture—including domain flexibility, Fc–Fab orientation, and IgG subclass—plays an important role in determining steric hindrance, molecular crowding, and self-interaction propensity, which ultimately influence viscosity behavior in high-concentration mAb formulations (35, 38, 39).
Protein–excipient interactions
• Sugars and polyols: These excipients increase solvent viscosity and promote preferential hydration of proteins, which can reduce protein–protein interactions and improve solution stability.
• Amino acids (arginine, histidine, lysine): Amino acids may disrupt reversible self-association by interacting with charged or aromatic residues on the antibody surface, thereby weakening intermolecular interactions.
• Surfactants (polysorbates, poloxamers): Surfactants primarily stabilize proteins at interfaces and can modulate interfacial rheology; however, their ability to reduce viscosity is often limited in highly concentrated antibody solutions.
Protein–excipient interactions, mediated by sugars, amino acids, and surfactants, can reduce self-association and interfacial stresses in antibody formulations. However, their effect on viscosity is formulation-dependent and often limited at very high protein concentrations (40–42).
Water and solvent structure effects
• Hydration shell dynamics: At high protein concentrations, hydration layers around antibodies begin to overlap. This reduces solvent mobility and creates energetic constraints for molecular rearrangement, contributing to increased solution viscosity.
• Crowding effects: High protein concentration also leads to excluded volume effects, which increase intermolecular collision frequency and promote transient clustering.
Water structuring and molecular crowding, driven by overlapping hydration shells and excluded volume effects, reduce solvent mobility and enhance intermolecular interactions, thereby contributing to increased viscosity in high-concentration mAb formulations (43–45).
Key contributors include electrostatic, hydrophobic, and aromatic/cation–π interactions, glycosylation patterns, structural architecture, and solvent/crowding effects, all of which drive reversible self-association and nonlinear viscosity increases. Protein–excipient interactions provide partial mitigation, but high concentrations (>100–150 mg/mL) remain prone to rheological and stability challenges.
Molecular engineering approaches to reduce viscosity
Molecular engineering strategies can help reduce viscosity in high-concentration monoclonal antibody (mAb) formulations by modifying surface properties that drive protein–protein interactions. Charge optimization through targeted mutations can redistribute surface charge and increase electrostatic repulsion between antibody molecules, thereby reducing reversible self-association and lowering viscosity (36).
Another approach involves reducing exposed hydrophobic or aromatic surface patches, which are known to promote non-specific intermolecular contacts. Substituting these residues with more hydrophilic amino acids can weaken attractive interactions and improve solution rheology without affecting antigen binding (38).
In addition, Fc engineering and structural modifications can influence antibody conformation and Fc–Fab orientation, thereby altering steric interactions and reducing intermolecular clustering in concentrated solutions (28). These molecular engineering strategies, combined with formulation optimization, provide effective approaches to mitigate viscosity challenges in high-concentration mAb products.
Predicting and measuring viscosity
Biophysical screening approaches
Early-stage viscosity assessment increasingly relies on low-material screening assays that provide rapid developability indicators while conserving valuable protein samples. Dynamic and static light scattering (DLS/SLS) are widely used to estimate the diffusion interaction parameter (kD) and the second virial coefficient (B22), both of which serve as surrogate indicators of protein–protein interactions in dilute solutions (46). Sedimentation velocity analytical ultracentrifugation (SV-AUC) further enables quantification of reversible self-association across concentration regimes by resolving oligomeric species and interaction equilibria (47). In addition, micro-rheology techniques and microfluidic capillary viscometers allow estimation of viscosity using sub-milligram quantities of protein, making them valuable tools during early formulation screening (32).
Although correlations between kD, B22, and solution viscosity are useful for early candidate ranking, extrapolation from dilute-solution measurements often fails to capture the steep, nonlinear viscosity increases observed at ultra-high protein concentrations (>150 mg/mL). At these concentrations, additional factors such as macromolecular crowding, hydration shell overlap, and non-ideal solution thermodynamics dominate rheological behavior (48).
Emerging technologies, including microfluidic screening platforms and automated high-throughput formulation systems, are increasingly being applied to accelerate viscosity characterization. Microfluidic rheometers measure flow resistance in microchannels to estimate viscosity under controlled shear conditions using minimal sample volumes. In parallel, high-throughput formulation platforms combined with automated liquid handling enable rapid screening of buffer composition, pH, ionic strength, and excipients to identify viscosity-reducing conditions. Integration of these platforms with data analytics and computational tools provides a powerful framework for rapid formulation optimization and early developability assessment of high-concentration antibody candidates (49).
Computational and machine learning models
Recent years have seen rapid progress in in-silico prediction of viscosity in high-concentration mAb formulations. Sequence-based ML models utilize features such as amino-acid composition, charge distribution, hydrophobicity indices, and predicted isoelectric point to identify viscosity liabilities early during antibody discovery (50). Structure-aware frameworks extend these approaches by incorporating three-dimensional antibody models, electrostatic surface potentials, and hydrophobic patch descriptors, which improve mechanistic interpretation of intermolecular interactions (51).
More advanced multi-modal ML architectures integrate sequence embedding with physicochemical developability descriptors such as hydrophobic surface area, charge asymmetry, dipole moment, and experimental interaction parameters (e.g., kD, B22, or opalescence measurements) to predict viscosity across wide concentration ranges (52). These approaches are increasingly combined with physics-informed models and colloidal interaction theories to better extrapolate viscosity behavior to ultra-high concentrations (>200 mg/mL), where conventional dilute-solution correlations often fail (53).
Recent molecular dynamics (MD) simulation studies have further provided mechanistic insight into protein–protein interactions in concentrated antibody solutions. MD simulations reveal how anisotropic charge distributions, hydrophobic surface patches, and dipole–dipole interactions promote transient clustering and reversible self-association networks that contribute to nonlinear viscosity increases at high protein concentrations (54). In addition, coarse-grained MD and Brownian dynamics simulations have been used to explore crowding effects, hydration layer overlap, and orientation-dependent antibody interactions, helping to explain the emergence of complex rheological behavior in highly concentrated systems (55).
The predictive performance of computational models improves with large and diverse antibody training datasets, rigorous cross-validation strategies, and explicit uncertainty quantification, which help identify prediction reliability and guide experimental validation. As a result, computational modeling and AI-driven approaches are becoming increasingly valuable tools for early developability assessment, rational antibody engineering, and formulation optimization in high-concentration mAb development.
Good measurement and reporting practices
To ensure translational relevance, viscosity characterization should follow several key best practices. First, measurements should be performed across the full intended concentration range, rather than relying solely on dilute or intermediate regimes where protein–protein interactions are less pronounced. Second, studies should report contextual formulation variables, including temperature, pH, ionic strength, and excipient composition, as these parameters strongly influence intermolecular interactions and rheological behavior (38). Third, viscosity measurements should incorporate shear rate conditions representative of real-world administration devices, such as autoinjectors or wearable pumps, to capture the non-Newtonian shear-thinning behavior commonly observed in concentrated monoclonal antibody (mAb) solutions (56).
In addition, computational predictions should be complemented with orthogonal experimental assays, including microfluidic rheometry, syringeability testing, and injection-force measurements, to ensure reliable characterization of formulation performance under clinically relevant conditions. Integration of these experimental and modeling approaches enables early identification of viscosity liabilities, reduces formulation optimization cycles, and improves confidence in candidate selection for high-concentration antibody development (32). Table 2 and Figure 2 summarize approaches for predicting and measuring viscosity in high-concentration mAbs.
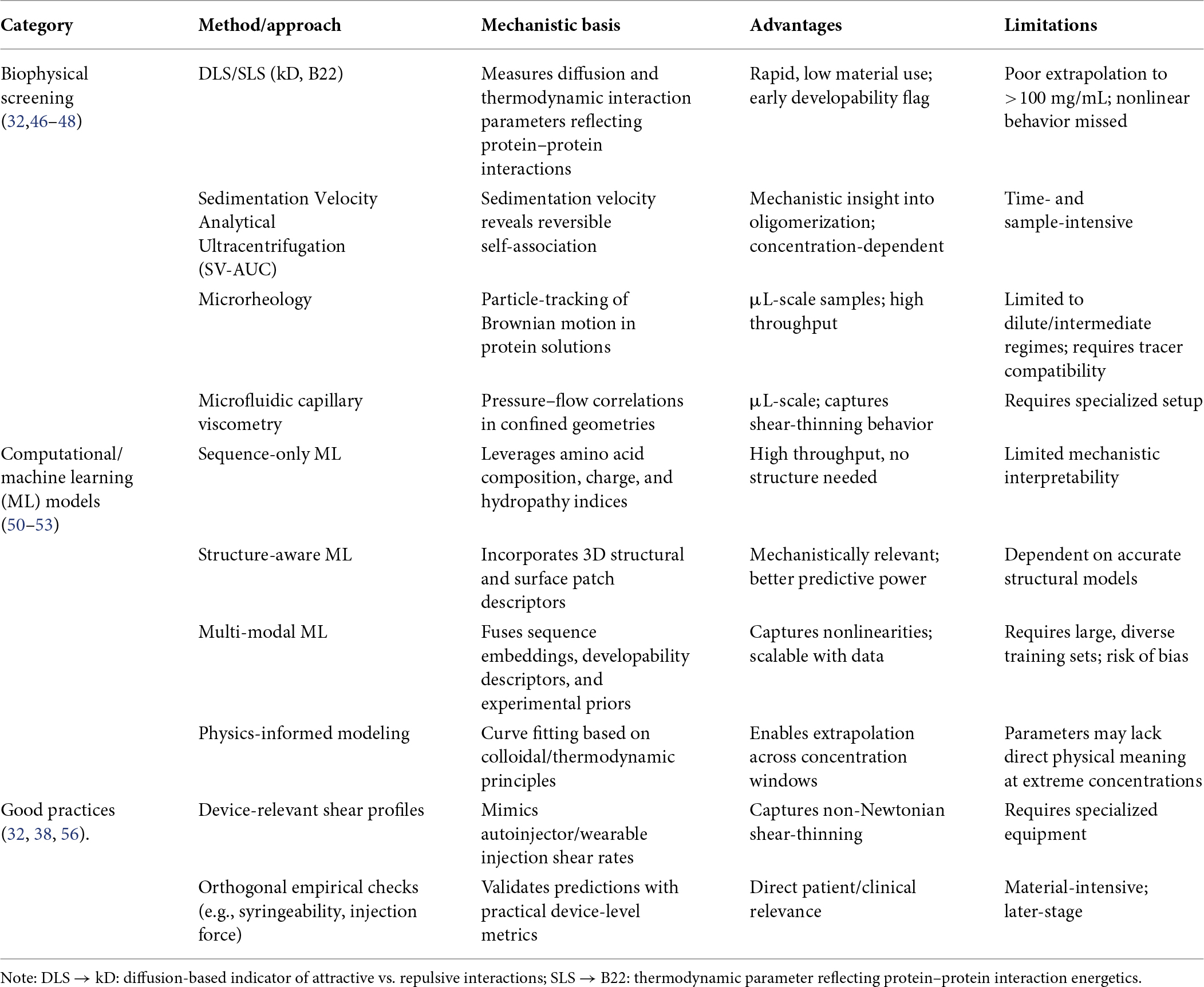
Table 2. Approaches for predicting and measuring viscosity in high-concentration monoclonal antibodies (mAbs).
Figure 2. Workflow for predicting and measuring viscosity in high-concentration monoclonal antibody formulations.
Strategies to reduce viscosity while preserving stability
Reducing viscosity in high-concentration monoclonal antibody (mAb) formulations requires an integrated strategy that balances intermolecular interaction control with preservation of conformational stability. Approaches typically involve solution conditioning, excipient selection, molecular engineering, and optimized processing conditions. These strategies aim to attenuate reversible protein–protein interactions that drive viscosity while maintaining long-term stability and manufacturability (39).
Solution conditioning
pH and ionic strength adjustment: Adjusting formulation pH away from the antibody’s isoelectric point (pI) increases net charge and enhances electrostatic repulsion, thereby reducing reversible self-association. Ionic strength can further modulate intermolecular interactions; salts such as NaCl or arginine hydrochloride can selectively screen attractive charge–patch interactions and lower viscosity. However, excessive ionic strength may destabilize protein structure or promote aggregation through charge neutralization, requiring careful optimization (57).
Buffer selection: Buffer identity influences both physicochemical stability and clinical tolerability. Histidine and acetate buffers are frequently used in SC formulations due to their buffering capacity in the physiological pH range. In contrast, citrate buffers may cause injection-site discomfort due to calcium chelation. Buffer composition can also influence pH drift, oxidation, and aggregation under stressed storage conditions.
Temperature management: Temperature affects the balance between enthalpic and entropic contributions governing protein–protein interactions. Elevated temperatures during manufacturing, shipping, or storage may enhance reversible clustering and viscosity. Consequently, low-temperature processing and controlled cold-chain logistics are often required to maintain acceptable rheological behavior in concentrated formulations.
Excipients and additives
Amino acids and ionic liquids: Certain amino acids such as arginine, lysine, histidine, and proline derivatives act as viscosity modifiers by interacting with charged or aromatic residues on the antibody surface. Arginine in particular disrupts electrostatic and π–π interactions through preferential binding to surface patches and modulation of solvent structure, thereby reducing intermolecular association (58). Emerging studies also explore ionic-liquid-like cosolutes and amino-acid-based ionic liquids as viscosity-reducing agents that alter hydration dynamics and intermolecular potentials while maintaining protein stability.
Sugars and polyols: Disaccharides such as sucrose and trehalose, as well as polyols such as glycerol, stabilize proteins through preferential hydration mechanisms, which favor the folded state and reduce attractive protein–protein contacts. In lyophilized systems, however, excipients such as mannitol may crystallize and affect cake structure or reconstitution behavior (59).
Surfactants: Surfactants such as polysorbate 20 (PS20), polysorbate 80 (PS80), and poloxamers primarily protect proteins from interfacial-induced aggregation during processing and administration. Although they have limited direct effects on bulk viscosity, they play an essential role in stabilizing formulations during pumping, filling, and injection. At high protein concentrations, degradation products of polysorbates (e.g., peroxides or free fatty acids) may introduce additional stability risks that require monitoring (60).
Novel cosolute systems: Recent formulation approaches employ multi-component cosolute systems, including betaine derivatives, proline analogs, or mixed osmolytes, which can reduce viscosity by modulating solvent structure and weakening protein–protein interactions. These strategies may achieve significant viscosity reductions (>30%) but require careful toxicological evaluation and regulatory justification for clinical use.
Protein engineering
Surface redesign: Computational and experimental approaches can identify interaction hotspots, such as hydrophobic patches or charge-complementary regions in the variable domain. Targeted mutations that redistribute surface charge or reduce hydrophobic exposure can weaken reversible self-association while preserving antigen binding and biological activity (36).
Glycoengineering and isotype selection: Fc glycosylation patterns influence steric hindrance and intermolecular packing. Controlled glycan remodeling or strategic IgG subclass selection (e.g., IgG2 vs. IgG1/IgG4) can reduce Fc-mediated clustering and improve rheological behavior while maintaining required effector functions.
Process and presentation
Concentration pathways: Downstream processing must minimize shear and interfacial stress during concentration. Tangential flow filtration (TFF) with optimized membrane selection and flux conditions helps prevent aggregation during ultrafiltration and diafiltration. Sequencing of excipient addition is also important to avoid local supersaturation or transient pH shifts that may trigger association.
Presentation format: Both liquid and lyophilized presentations are used for high-concentration mAb products. In lyophilized formulations, reconstitution to high concentration requires careful management of opalescence, foaming, and reconstitution forces. Excipient selection and lyophilization cycle design strongly influence reconstitution time, solution clarity, and syringeability.
Amino acids and ionic liquids as viscosity modifiers
Amino acids are widely used as viscosity-modifying excipients in high-concentration monoclonal antibody (mAb) formulations due to their ability to modulate intermolecular interactions. Among these, arginine, lysine, histidine, and proline derivatives are particularly effective in reducing solution viscosity. Arginine acts through multiple mechanisms, including electrostatic screening, disruption of aromatic π–π interactions, and preferential interactions with hydrophobic surface patches on antibody molecules. These interactions weaken reversible self-association and reduce the formation of transient intermolecular networks responsible for high viscosity (40). Arginine is often used alone or in combination with glutamate (arginine–glutamate mixtures), which further enhances viscosity reduction by modulating solvent structure and altering the effective interaction potential between antibody molecules. Lysine and histidine can also interact with charged residues on antibody surfaces, thereby reducing attractive charge–patch interactions and improving rheological behavior in concentrated formulations (61).
More recently, ionic liquids and amino-acid–based ionic liquids have emerged as novel cosolutes capable of modulating protein–protein interactions. These systems influence hydration shell structure, electrostatic screening, and hydrogen-bonding networks, which can reduce intermolecular attraction and stabilize protein solutions at high concentrations. Certain ionic liquid systems have demonstrated significant viscosity reductions while maintaining conformational stability and minimizing aggregation, although their use requires careful consideration of toxicological, pharmacokinetic, and regulatory factors in therapeutic formulations (62).
Overall, amino acids and ionic-liquid-based excipients represent promising tools for fine-tuning intermolecular interactions and reducing viscosity in high-concentration mAb formulations, particularly when used in combination with optimized buffer conditions and other stabilizing excipients (63). Table 3 and Figure 3 summarize strategies and approaches to reduce viscosity in high-concentration mAbs while preserving stability, respectively.
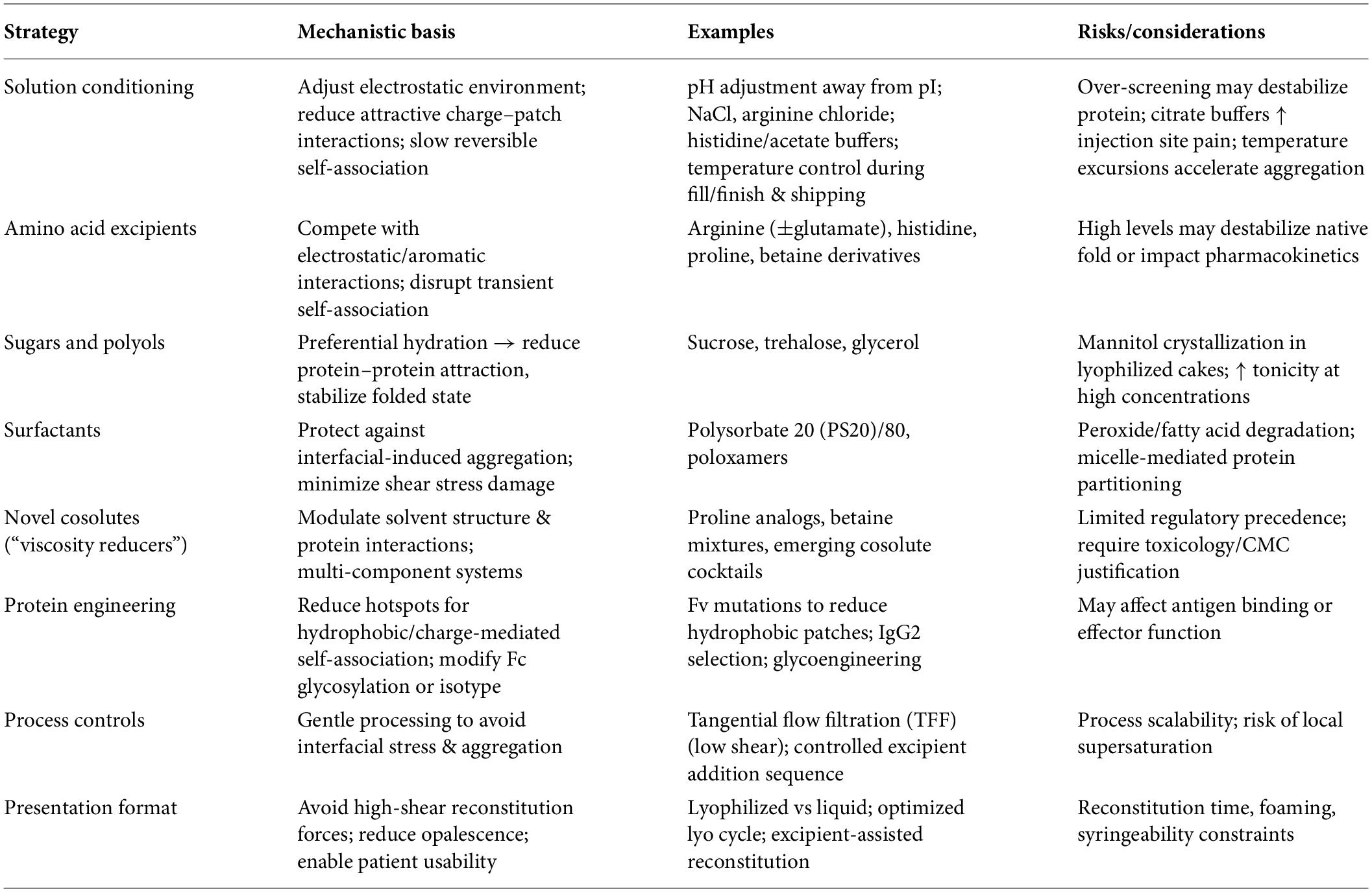
Table 3. Strategies to reduce viscosity in high-concentration mAbs while preserving stability.
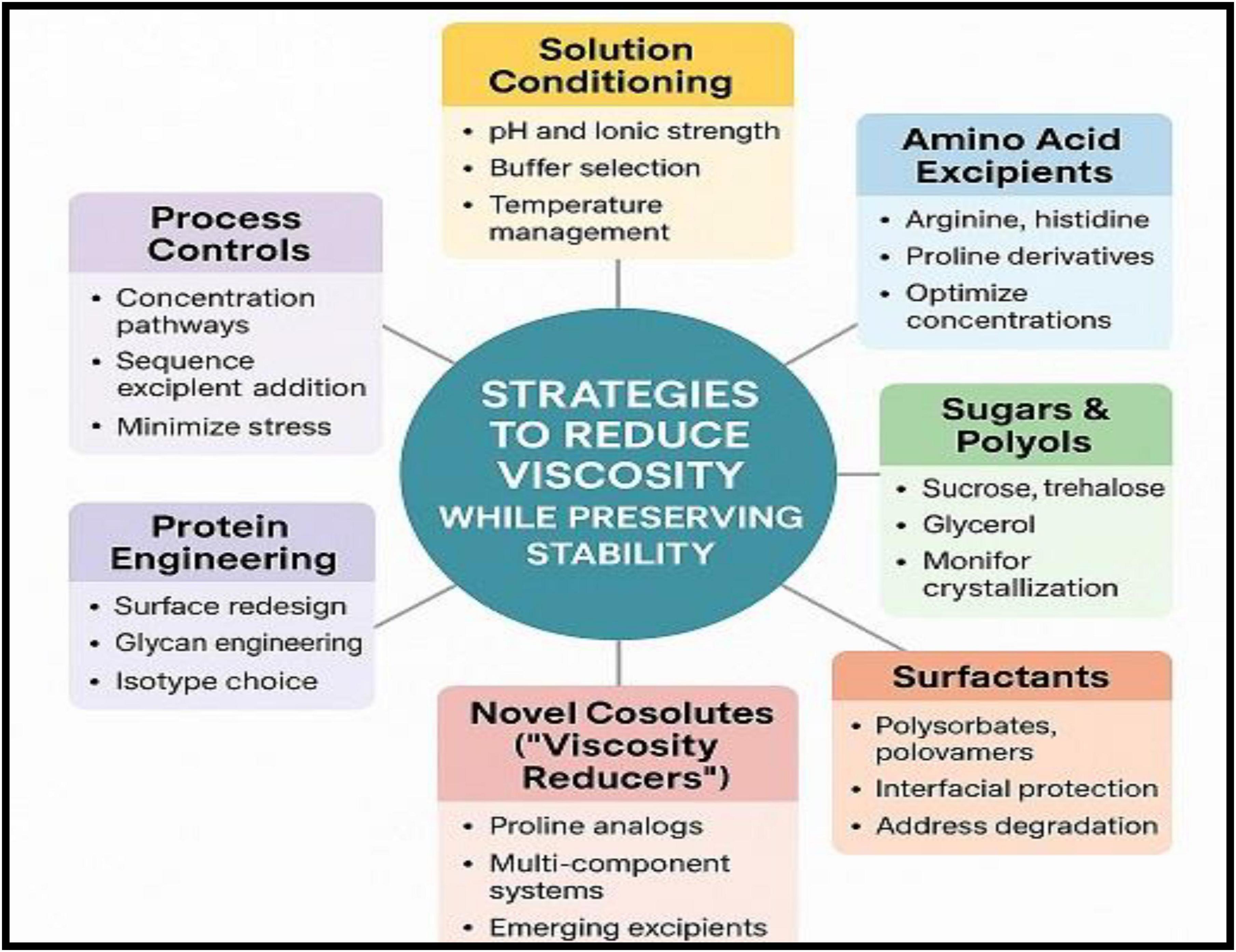
Figure 3. Strategies to reduce viscosity in high-concentration monoclonal antibody formulations while preserving stability.
Subcutaneous delivery, large-volume subcutaneous (LVSC) administration, and enabling technologies
Device constraints
Traditional SC delivery of mAbs is constrained by both physiological limitations of the SC tissue and the mechanical capabilities of injection devices. Conventional autoinjector systems are typically limited to injection volumes of approximately 1–2 mL, while maintaining acceptable injection forces and administration times to ensure patient comfort and usability (22). These constraints place practical limits on the concentration and viscosity of mAb formulations intended for SC administration.
Recent advances in wearable on-body delivery systems (OBDS) have expanded the feasible dosing volume for SC administration. These systems can deliver larger volumes (approximately 3–10 mL) over extended infusion durations ranging from 5 to 30 minutes, thereby enabling administration of higher therapeutic doses without the need for IV infusion (51). Such devices are increasingly used for LVSC delivery of biologics.
The rheological properties of high-concentration antibody formulations play a critical role in device compatibility. Many mAb solutions exhibit shear-thinning behavior, where viscosity decreases under high shear conditions encountered during injection through narrow needles or micro-tubing. However, zero-shear viscosity remains the key determinant governing flow resistance, injection force, and device performance (64). Consequently, formulation development must carefully balance protein concentration, viscosity, and device specifications to maintain acceptable injection forces and ensure reliable drug delivery.
In addition to rheological considerations, device compatibility also requires evaluation of needle gauge, flow rate, injection time, and patient usability factors, all of which influence overall treatment acceptance and adherence. Integration of formulation optimization with device engineering is therefore essential for successful development of SC and LVSC antibody therapies (65).
Hyaluronidase-enabled delivery
Recombinant human hyaluronidase (rHuPH20) has emerged as a clinically validated strategy to expand the SC delivery window for biologics. rHuPH20 temporarily depolymerizes hyaluronan within the extracellular matrix of SC tissue, thereby reducing tissue resistance and increasing local permeability. This transient remodeling of the interstitial matrix enables administration of larger injection volumes at faster infusion rates, facilitating LVSC delivery of mAbs that would otherwise require IV administration (66).
Several marketed biologics including trastuzumab, rituximab, and daratumumab are now available in co-formulated or co-administered rHuPH20 products, demonstrating the clinical feasibility, safety, and patient acceptance of this approach (67). These formulations enable delivery of multi-milliliter doses through SC injection while maintaining pharmacokinetic exposure comparable to IV administration.
Beyond currently approved products, ongoing development programs are exploring rHuPH20-enabled LVSC administration for additional mAbs and biologics, particularly those requiring high therapeutic doses or frequent infusion schedules. This strategy has the potential to improve patient convenience, reduce infusion times, and shift treatment from hospital-based IV administration to outpatient or home settings (63).
Regulatory precedents established by approved rHuPH20-containing products provide a clear pathway for future development of enzyme-enabled SC biologic therapies. Nevertheless, successful implementation requires careful attention to co-formulation stability, enzyme activity control, compatibility with device systems, and long-term product stability, all of which must be addressed during formulation and process development (68).
Formulation–device co-optimization
Successful SC and LVSC delivery requires coordinated optimization of both formulation properties and device mechanics. Key device parameters—including needle gauge, drive force, plunger mechanics, and spring or motor capacity—must be carefully matched with formulation rheology, particularly viscosity and shear-thinning behavior. High-concentration monoclonal antibody (mAb) formulations often exhibit non-Newtonian flow characteristics, and therefore device design must accommodate the injection forces required to deliver viscous solutions through narrow needles within acceptable administration times (69).
In addition to mechanical compatibility, patient-centric considerations play an essential role in SC product development. Factors such as injection-site pain, injection duration, and overall usability can significantly influence treatment adherence. Injection-site discomfort has been associated with formulation attributes including buffer composition, osmolality, and pH, highlighting the importance of integrating formulation design with device performance requirements (70).
Regulatory agencies increasingly encourage holistic, risk-based development strategies that consider formulation attributes, delivery device performance, and patient usability within a unified control framework. Such integrated approaches align with QbD principles, emphasizing systematic evaluation of critical quality attributes (CQAs), device functionality, and user experience during product development (71). This paradigm underscores the need for interdisciplinary collaboration among formulation scientists, device engineers, and clinical teams to enable safe, reliable, and patient-acceptable SC and LVSC dosing of biologic therapies.
Stability challenges at high concentration
Physical instability
High-concentration monoclonal antibody (mAb) formulations are particularly susceptible to physical instability arising from enhanced protein–protein interactions and molecular crowding. At elevated concentrations, reversible self-association between antibody molecules can lead to the formation of transient oligomers or clusters, which may subsequently nucleate irreversible aggregation or gel-like network formation under storage or stress conditions (72). These processes are often driven by electrostatic, hydrophobic, and dipole–dipole interactions that become increasingly significant as intermolecular distances decrease.
Liquid–liquid phase separation (LLPS) is another important phenomenon observed in concentrated protein solutions. In LLPS, the formulation separates into a protein-rich and protein-poor phase, typically triggered by attractive intermolecular interactions or changes in temperature, pH, or ionic strength. This behavior can significantly alter viscosity and protein stability while complicating manufacturability and product appearance (73). Closely related phenomena such as opalescence or reversible clustering may occur at concentrations approaching the LLPS boundary and can create challenges for visual inspection and quality control.
Interfacial stresses also play a major role in aggregation pathways. Exposure of proteins to air–liquid, liquid–solid, or liquid–oil interfaces during processing, shipping, agitation, or device priming can induce partial protein unfolding and adsorption at interfaces. Subsequent desorption of these destabilized species often leads to particle formation and accelerated aggregation in solution (74). Such effects are particularly relevant for high-concentration formulations, where even small perturbations may initiate aggregation cascades.
Furthermore, the presence of high solution viscosity and crowding effects can slow molecular diffusion and promote local concentration heterogeneity, increasing the probability of intermolecular collisions and aggregation events. These phenomena collectively complicate visual inspection and pharmacopeial requirements for clarity and particulate matter control, especially in formulations intended for SC administration (14, 75).
Chemical pathways
In high-concentration monoclonal antibody (mAb) formulations, molecular crowding and increased microviscosity can influence the kinetics and pathways of chemical degradation relative to dilute systems. Restricted diffusion and altered solvent dynamics may increase local residence times of reactive species, thereby accelerating certain chemical modifications such as deamidation, oxidation, and glycation.
Deamidation is one of the most common degradation pathways in antibodies and typically occurs at asparagine (Asn) residues, particularly within flexible regions such as CDRs. The reaction proceeds through the formation of a succinimide intermediate, which subsequently hydrolyzes to yield aspartate (Asp) or isoaspartate (isoAsp) residues. This modification can alter local charge distribution, disrupt hydrogen bonding networks, and potentially affect antigen-binding affinity or structural stability. Deamidation rates are influenced by pH, temperature, local sequence context (e.g., Asn–Gly motifs), and conformational flexibility and may be enhanced in crowded environments where local protein–protein interactions alter solvent accessibility.
Oxidation represents another important chemical instability pathway and primarily affects methionine (Met), tryptophan (Trp), histidine (His), and cysteine residues. Oxidative reactions are often initiated by reactive oxygen species (ROS) or trace peroxides present in formulation components. For example, oxidation of Met residues within the Fc region can modify Fc receptor interactions and impact biological activity. Oxidation of Trp residues may also disrupt tertiary structure and promote aggregation. In high-concentration formulations, oxidation risk may be amplified due to localized accumulation of reactive species and reduced diffusion of antioxidants or scavengers.
Surfactants such as PS20 or PS80 are commonly included in formulations to prevent interfacial aggregation; however, their degradation products can introduce additional oxidative stress. Polysorbates may undergo auto-oxidation, generating peroxides and free fatty acids that can initiate oxidation reactions in proteins. Consequently, strict control of excipient purity, peroxide levels, oxygen exposure, and storage conditions is essential to minimize oxidative degradation in concentrated mAb formulations (76–78).
Overall, chemical degradation pathways such as deamidation and oxidation can compromise protein structure, biological activity, and long-term stability and therefore must be carefully monitored and controlled during formulation development and storage of high-concentration antibody products.
Container–closure interactions
Prefilled syringes and autoinjector systems introduce additional container–closure–related stability risks in high-concentration monoclonal antibody (mAb) formulations. Silicone oil is commonly applied as a lubricant on syringe barrels to ensure smooth plunger movement; however, silicone oil microdroplets can promote protein adsorption at oil–water interfaces and act as nucleation sites for aggregate formation or visible/sub-visible particles (79). In highly concentrated protein solutions, such interfacial interactions may accelerate aggregation or contribute to particle formation during storage, transport, or device actuation.
Manufacturing residues can also influence product stability. For example, trace tungsten residues originating from syringe needle drilling processes may leach into the formulation and promote protein aggregation or chemical modification through localized pH shifts or catalytic effects (59). Such interactions have been associated with destabilization of certain protein formulations and require careful control of syringe manufacturing processes.
In addition, extractables and leachables originating from polymeric device components—such as elastomeric plungers, adhesives, or plastic housings—may interact with antibody molecules, potentially affecting stability, potency, or immunogenicity risk. Regulatory agencies, therefore, emphasize comprehensive container–closure compatibility assessments, including identification and qualification of potential leachables.
To address these risks, current regulatory guidance recommends risk-based control strategies supported by orthogonal analytical methods. Techniques such as sub-visible particle analysis (light obscuration), micro-flow imaging (MFI), resonant mass measurement (RMM), and spectroscopic characterization are widely used to detect particles and monitor formulation stability during development and storage (51). These analytical approaches enable early identification of container–closure-related instabilities and support the establishment of robust control strategies to ensure product quality throughout the product lifecycle. Table 4 summarizes stability challenges in high-concentration mABs.
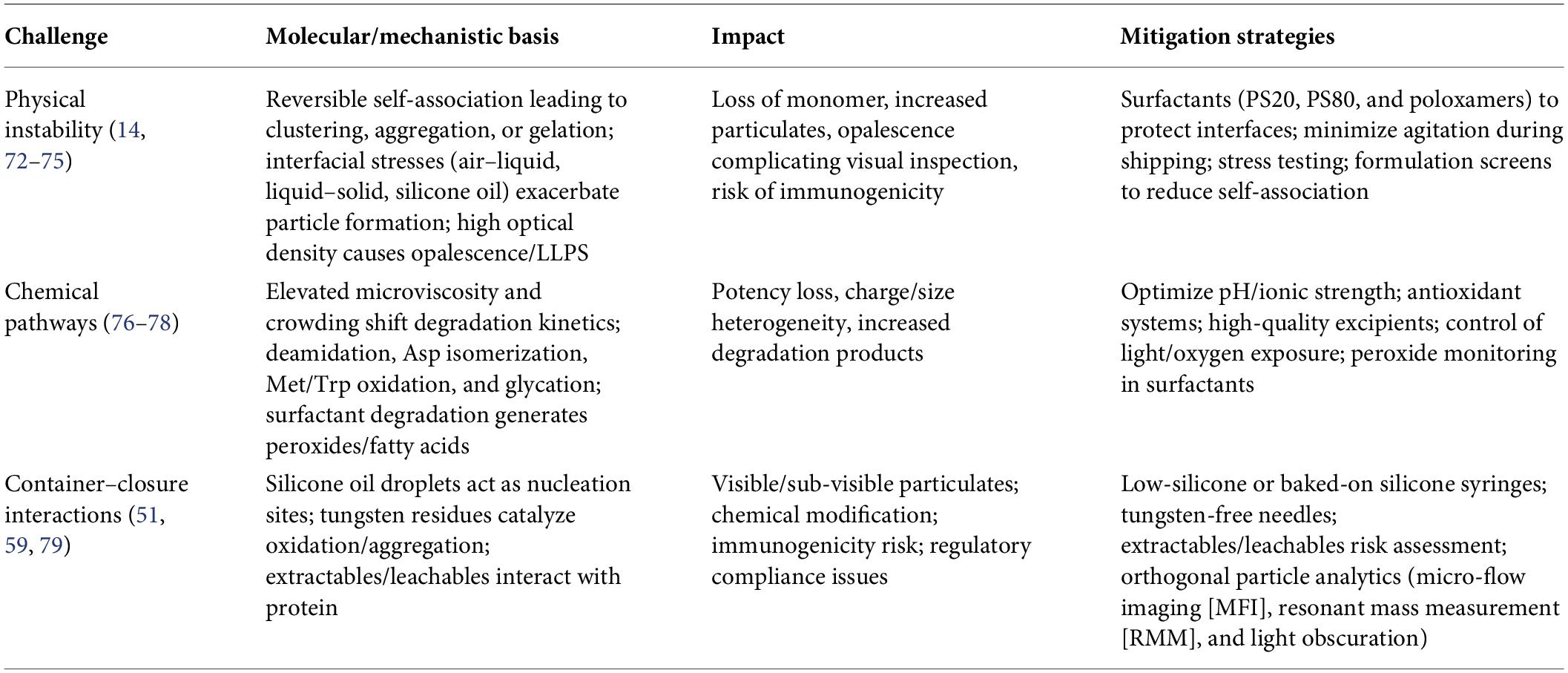
Table 4. Stability challenges in high-concentration monoclonal antibody formulations.
Impact of pH, ionic strength, and excipients on stability
The stability of high-concentration monoclonal antibody (mAb) formulations is strongly influenced by pH, ionic strength, and excipient composition, which collectively regulate protein conformation, intermolecular interactions, and chemical degradation pathways.
Formulation pH plays a critical role in determining the net surface charge of antibodies relative to their isoelectric point (pI). Maintaining formulation pH sufficiently away from the pI increases electrostatic repulsion between molecules, thereby reducing reversible self-association, aggregation, and viscosity. In contrast, formulations near the pI may promote attractive protein–protein interactions and increase susceptibility to aggregation and phase separation (25).
Ionic strength also modulates protein stability by influencing electrostatic interactions and shielding charged residues on the antibody surface. Moderate salt concentrations can stabilize proteins by screening unfavorable charge–patch interactions, whereas excessive ionic strength may reduce repulsive forces and promote aggregation or structural destabilization. Consequently, careful optimization of ionic strength is required to balance stability and viscosity in concentrated antibody formulations.
Excipients further contribute to stabilization by influencing both protein conformation and solvent structure. Sugars and polyols such as sucrose, trehalose, and glycerol stabilize antibodies through preferential hydration, favoring the native folded state and reducing aggregation propensity. Amino acids such as arginine and histidine can suppress reversible self-association by interacting with protein surface residues and modifying solvent properties. Surfactants, including PS20 and PS80, primarily protect proteins from interfacial stresses during manufacturing and storage, thereby reducing aggregation triggered by surface adsorption (80).
Collectively, the careful selection and optimization of pH, ionic strength, and stabilizing excipients are essential for maintaining conformational stability, minimizing aggregation, and ensuring long-term stability of high-concentration mAb formulations.
Quality by Design (QbD) and risk management in high-concentration SC mAb formulations
Application of QbD principles is essential to ensure that high-concentration monoclonal antibody (mAb) formulations intended for SC or LVSC delivery are robust, manufacturable, and patient-centric. Unlike traditional empirical formulation approaches, QbD provides a systematic, science- and risk-based framework that integrates product understanding, process control, and patient-use requirements throughout development (81). Table 5 shows a QbD outline for high-concentration SC mAb formulations.

Table 5. QbD framework for high-concentration subcutaneous (SC) mAb formulations.
The QbD process begins with defining the Quality Target Product Profile (QTPP), which outlines the desired clinical and product performance characteristics. For SC mAb formulations, the QTPP typically includes dose strength, target protein concentration, injection volume limits, acceptable viscosity range, injection time targets, and compatibility with the intended delivery system (e.g., autoinjector, prefilled syringe, wearable injector, or LVSC platform) (82). Patient-centric attributes such as injection comfort, device usability, and administration setting (clinic vs. home use) are also considered during QTPP definition.
From the QTPP, CQAs are identified. For high-concentration antibody formulations, CQAs commonly include solution viscosity across relevant shear rates, aggregation and particulate levels, potency and biological activity, charge variants, size variants, and chemical stability markers such as oxidation or deamidation. These attributes directly influence product safety, efficacy, and usability (81). The next step involves identifying Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) that affect CQA variability. Important CMAs include formulation pH, ionic strength, buffer identity, excipient composition, and raw material quality, while CPPs may include ultrafiltration/diafiltration (UF/DF) concentration methods, shear exposure during pumping and filling, temperature conditions, and container–closure interactions. Understanding these relationships enables identification of process variables that must be controlled to ensure consistent product performance (83).
Risk assessment tools such as Failure Modes and Effects Analysis (FMEA) and Ishikawa (fishbone) diagrams are widely used to prioritize variables that have the greatest impact on product quality and patient usability. These tools allow visualization of potential risks associated with formulation composition, device interaction, processing conditions, and storage environment, thereby guiding experimental design and control strategies (84).
To establish a design space, multivariate Design of Experiments (DoE) studies are conducted to systematically evaluate interactions between key formulation variables. For example, factorial or response surface designs may examine the combined effects of pH, salt concentration, and excipient ratios on viscosity, colloidal stability, and aggregation propensity. Such structured experimentation enables identification of optimal formulation regions where product performance remains robust despite minor variations in processing conditions (85).
Compared with conventional trial-and-error approaches, QbD provides several advantages. It enables early identification of formulation and process risks, improved process understanding, and establishment of robust control strategies. In addition, regulatory agencies increasingly support QbD-based submissions because the framework demonstrates comprehensive product knowledge and facilitates lifecycle management. By linking formulation design, manufacturing processes, and device performance within an integrated control strategy, QbD reduces late-stage development failures and ensures consistent clinical performance of high-concentration SC mAb products.
Case studies in high-concentration SC mAb formulations
Case study 1: trastuzumab and rituximab SC (rHuPH20-enabled, Roche/Genentech)
Challenge: Standard IV formulations of trastuzumab and rituximab require infusion of relatively large volumes due to dosing requirements, resulting in prolonged administration times in clinical settings.
Solution: Co-formulation with rHuPH20 enabled delivery of larger SC volumes (approximately 5–15 mL) by transiently increasing tissue permeability, allowing SC administration within a few minutes.
QbD Elements: The QTPP focused on reducing infusion burden while maintaining comparable pharmacokinetics to IV dosing. CQAs included viscosity, potency, stability of the dual-protein formulation, and syringeability. Risk mapping identified rHuPH20 stability and excipient compatibility as key CMAs.
Outcome: Approved products (Herceptin SC® and MabThera SC®) demonstrated non-inferior pharmacokinetics compared with IV formulations, while significantly improving patient convenience and reducing clinic chair time (69).
Case study 2: daratumumab SC (Janssen, Darzalex Faspro®)
Challenge: IV daratumumab infusions can require several hours, particularly during initial dosing, due to infusion-related reactions and the need for slow infusion rates.
Solution: A SC co-formulation with rHuPH20 enabled delivery of a fixed 1,800 mg dose in approximately 3–5 minutes, greatly simplifying administration.
QbD Approach: Development focused on CQAs, including viscosity (<20 cP), stability of co-formulated proteins, and compatibility with SC delivery devices. DoE was applied to optimize pH, buffer composition, and excipient balance to maintain stability and acceptable viscosity.
Outcome: The SC formulation (Darzalex Faspro®) received regulatory approval and demonstrated substantially reduced administration time and improved patient-reported outcomes compared with IV infusion (86).
Case study 3: adalimumab high-concentration, citrate-free formulations
Challenge: Early adalimumab formulations were 50 mg/mL citrate-buffered solutions, associated with relatively larger injection volumes and injection-site discomfort.
Solution: Reformulation to 100 mg/mL citrate-free products reduced injection volume and improved patient tolerability. Histidine buffers and optimized excipient systems were used to maintain stability while controlling viscosity.
QbD Focus: The QTPP emphasized improved patient comfort and reduced injection volume. Risk assessment tools such as FMEA identified buffer composition and ionic strength as key CMAs influencing injection pain and viscosity.
Outcome: Several marketed biosimilars and reformulated products achieved smaller injection volumes (≈0.4–0.5 mL) and improved patient preference for high-concentration SC dosing (87).
Case study 4: tocilizumab SC (Actemra®/RoActemra®)
Challenge: IV administration of tocilizumab required clinic-based infusions, limiting patient convenience and treatment accessibility.
Solution: Development of a high-concentration SC formulation (162 mg/0.9 mL) compatible with autoinjector devices enabled self-administration.
QbD Mapping: Key CQAs included solution viscosity (<15 cP), potency, aggregation control, and visual clarity. CPPs included UF/DF shear history and container–closure interactions, particularly silicone oil effects.
Outcome: The SC formulation enabled self-administration and expanded patient access to therapy while maintaining comparable safety and efficacy to IV administration (88).
Key learnings across case studies
Several important lessons emerge from these examples (70):
I. rHuPH20-enabled LVSC delivery significantly expands the feasible dosing range for SC biologics when viscosity or dose volume would otherwise require IV infusion.
II. High-concentration citrate-free reformulations improve injection tolerability and patient preference.
III. Quality by Design frameworks (QTPP → CQAs → CMAs/CPPs) enable systematic identification of formulation and process risks.
IV. Device–formulation co-optimization, including needle gauge, injection force, and wearable device compatibility, is essential for successful SC biologic development.
Additional commercial examples of high-concentration SC mAb formulations
Several additional monoclonal antibody products illustrate successful implementation of high-concentration SC formulations.
For example, dupilumab (Dupixent®) and secukinumab (Cosentyx®) are delivered as concentrated SC formulations using autoinjector or prefilled syringe devices. These products demonstrate how optimized buffer systems, viscosity control, and device compatibility enable convenient at-home administration of biologics requiring repeated dosing (89).
Similarly, ustekinumab (Stelara®) and omalizumab (Xolair®) highlight the importance of formulation stability and device integration for chronic therapy. These products rely on carefully optimized protein concentration, excipient systems, and injection device design to ensure acceptable viscosity, stability, and patient usability (90).
Conclusion
High-concentration monoclonal antibody formulations have enabled the transition from IV infusion to patient-friendly SC delivery, supporting self-administration and improved healthcare efficiency. However, achieving concentrations above 100–150 mg/mL introduces significant challenges related to viscosity, physical and chemical stability, manufacturability, and device compatibility. These challenges arise from complex molecular interactions—including electrostatic, hydrophobic, aromatic, and glycosylation-mediated effects—as well as solvent structuring and molecular crowding. Advances in biophysical screening, predictive modeling, and machine-learning approaches are improving the early identification of viscosity liabilities, while formulation strategies involving excipient optimization, protein engineering, and process control continue to enhance stability and manufacturability.
Despite these advances, important knowledge gaps remain. A deeper mechanistic understanding of protein–protein interactions at ultra-high concentrations (>200 mg/mL), improved predictive models that integrate molecular structure with formulation variables, and standardized rheological characterization methods are still needed. Emerging therapeutic modalities—including bispecific antibodies, antibody fragments, and nanobody-based biologics—may introduce additional formulation complexities due to distinct structural and interaction profiles. Future progress will depend on integrating molecular design, advanced excipient systems, AI-driven developability prediction, and device-formulation co-optimization within robust QbD frameworks.
Overall, the successful development of next-generation high-concentration antibody formulations will rely on interdisciplinary approaches that combine molecular engineering, formulation science, process optimization, and delivery technology, ultimately enabling more accessible, stable, and patient-centric biologic therapies.
Future outlook
Advances in understanding the molecular drivers of viscosity—including electrostatic, hydrophobic, and aromatic/cation–π interactions, as well as Fc glycosylation, antibody structural architecture, excipient interactions, and solvent structuring effects—are reshaping the design of high-concentration monoclonal antibody formulations. Future development is expected to increasingly emphasize rational protein engineering to minimize self-association hotspots, precision glycoengineering to control Fc sterics and heterogeneity, and computational and AI-driven predictive models that integrate sequence, structural, and formulation descriptors to identify viscosity liabilities early in development.
Formulation science will likely shift toward multi-component excipient systems, combining amino acids, sugars, and surfactants to synergistically control intermolecular interactions while preserving conformational and colloidal stability. In parallel, novel viscosity-modifying cosolutes and amino-acid–based excipient systems are emerging that may reduce solution viscosity by more than 30%–40% while maintaining long-term stability. Advances in solvent engineering and hydration-shell modulation may further expand the physicochemical design space for concentrated biologics by influencing crowding effects and intermolecular interaction potentials.
At the interface with drug delivery, device–formulation co-optimization and enzyme-enabled strategies such as rHuPH20 will remain central to expanding the feasibility of LVSC administration. Integration of wearable injection systems, advanced autoinjectors, and digital monitoring technologies may further enhance the usability and accessibility of biologic therapies. Embedding these innovations within QbD and risk-based development frameworks will help ensure regulatory alignment while reducing empirical formulation iteration.
Ultimately, the convergence of molecular design, predictive analytics, excipient innovation, and advanced delivery technologies will enable the next generation of high-concentration antibody therapeutics. These integrated approaches are expected to produce biologic products with improved stability, manufacturability, and patient usability, thereby supporting broader global access to antibody-based therapies.
List of abbreviations
mAb: monoclonal antibody
SC: subcutaneous
LVSC: large-volume subcutaneous
rHuPH20: recombinant human hyaluronidase PH20
LLPS: liquid–liquid phase separation
DLS: dynamic light scattering
SLS: static light scattering
AUC: analytical ultracentrifugation
SV-AUC: sedimentation velocity analytical ultracentrifugation
kD: diffusion interaction parameter
B22: second virial coefficient
Fc: fragment crystallizable region
Fab: fragment antigen binding region
IgG: immunoglobulin G
OBDS: on-body delivery system
Author contributions
Nirav Patel: Literature review, data collection, and manuscript drafting. Vaibhav Bhatt: Conceptualization, scientific review, and critical revision of the manuscript. Priya Patel: Conceptualization, supervision, manuscript writing, and final editing. All authors reviewed and approved the final manuscript.
Funding
The authors received no external funding for the preparation of this review article.
Acknowledgments
The authors acknowledge the use of artificial intelligence–assisted tools (CHATGPT, OPENAI) for draft preparation, language refinement, and conceptual illustration support during the development of this review article. All AI-assisted content was critically reviewed, validated, and edited by the authors to ensure scientific accuracy, technical integrity, and compliance with journal and academic standards.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
3. Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. mAbs. (2015) 7(1):9–14.
4. Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. Drug Discov Today. (2018) 23(6):1159–65.
5. Yu J, Wang J, Zhang Y, Chen G, Mao W, Ye Y et al. Glucose-responsive insulin patch for the regulation of blood glucose in mice and minipigs. J Control Release. (2020) 328:394–403.
7. Roberts CJ. Protein aggregation and its impact on product quality. Trends Biotechnol. (2014) 32(7):372–80.
8. Yadav S, Shire SJ, Kalonia DS. Factors affecting the viscosity in high concentration solutions of different monoclonal antibodies. Mol Pharm. (2018) 15(5):1865–75.
9. Liu J, Nguyen MDH, Andya JD, Shire SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci. (2019) 108(1):9–28.
10. Fesinmeyer RM, Hogan S, Saluja A, Brych SR, Kras E, Narhi LO et al. Effect of ions on agitation- and temperature-induced aggregation reactions of antibodies. J Pharm Sci. (2019) 108(9):2987–98.
11. Kinnunen HM, Mrsny RJ. Improving the outcomes of biopharmaceutical delivery via the subcutaneous route by understanding the chemical, physical and physiological properties of the subcutaneous injection site. Adv Drug Deliv Rev. (2014) 91:52–65.
12. Arakawa T, Ejima D, Li T, Tsumoto K. Suppression of protein interactions by arginine: a proposed mechanism of the arginine effects. Biotechnol Prog. (2006) 22(6):1547–54.
13. Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S et al. Rational design of viscosity-lowering mutations for monoclonal antibodies. J Pharm Sci. (2015) 104(2):394–403.
14. Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. (2004) 93(6):1390–402.
15. Yadav S, Laue TM, Kalonia DS, Singh SN, Shire SJ. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibodies in concentrated solutions. Mol Pharm. (2012) 9(4):791–802.
16. Roberts CJ, Das TK. Protein aggregation and viscosity in high-concentration antibody formulations. Trends Biotechnol. (2014) 32(7):372–80.
17. Barnett GV, Wang G, Li Y, Guo S, Wang B, Venkatraman V et al. Physicochemical determinants of viscosity in high concentration antibody solutions. Mol Pharm. (2020) 17(12):4421–32.
18. Scherer TM, Liu J, Ruan Q, Li Y, Liu D, Apostol I et al. Mechanisms of viscosity modulation of high concentration antibody solutions by arginine glutamate. J Pharm Sci. (2023) 112(2):431–48.
19. Mason BD, Zhang L, Remmele RL Jr., Zhang J. Opalescence of an IgG2 monoclonal antibody solution as it relates to liquid–liquid phase separation. mAbs. (2017) 9(4):682–95.
20. Yang Y, Xu T, Yu X, Sun Y, Li F, Zhao C et al. Understanding the high viscosity of concentrated monoclonal antibody solutions: Structural and interactional insights. Nat Commun. (2022) 13:4089.
21. Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS et al. Computational tool for the early screening of monoclonal antibodies for developability issues. J Pharm Sci. (2016) 105(9):2764–73.
22. Bittner B, Schmidt J, Tillmann C. Subcutaneous delivery of biotherapeutics: challenges and opportunities. Adv Drug Deliv Rev. (2021) 172:91–107.
23. Nurse R, Zepeda ML, Jose G, Chan L, Nunez DJ, Greenwood J. Clinical pharmacology of monoclonal antibodies for subcutaneous delivery. Clin Pharmacol Ther. (2020) 108(5):928–41.
24. Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: current experience in animal models and humans. J Pharm Sci. (2018) 107(4):1104–17.
25. ICH.ICH harmonised guideline Q8(R2): Pharmaceutical development. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (2009).
26. U.S. Food and Drug Administration.Considerations for the use of artificial intelligence to support regulatory decision-making for drug and biological products. Draft guidance for industry. FDA (2025).
27. Yadav S, Liu J, Shire SJ, Kalonia DS. Specific interactions in high-concentration protein solutions resulting in high viscosity. Mol Pharm. (2010) 7(3):851–62.
28. Liu J, Nguyen MD, Andya JD, Shire SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci. (2005) 94(9):1928–40.
29. Esfandiary R, Parupudi A, Casas-Finet J, Gadre D, Sathish HA. Mechanism of reversible self-association of a monoclonal antibody: role of electrostatics and contribution to solution viscosity. J Pharm Sci. (2015) 104(2):577–86.
30. Agrawal NJ, Helk B, Kumar S, Mody R, Sathish HA. Computational and experimental approaches for predicting viscosity of concentrated protein solutions: a review. J Pharm Sci. (2016) 105(9):2764–73.
31. Nichols P, Li L, Kumar S, Buck PM, Singh SK. Rational design of antibody molecules to lower solution viscosity. mAbs. (2015) 7(2):212–20.
32. Barnett GV, Razinkov VI, Kerwin BA, Liu D, Roberts CJ, Fernandez EJ et al. High-concentration antibody solutions: direct correlation between rheological properties and colloidal interactions. Mol Pharm. (2020) 17(12):4421–32.
33. Scherer TM, Liu J, Shire SJ, Carpenter JF. Mapping molecular determinants of viscosity in concentrated protein solutions. J Pharm Sci. (2023) 112(2):431–48.
34. Yadav S, Shire SJ, Kalonia DS. Factors affecting the viscosity in high-concentration solutions of different monoclonal antibodies. J Pharm Sci. (2010) 99(12):4812–29.
35. Roberts CJ. Therapeutic protein aggregation: mechanisms, design, and control. Trends Biotechnol. (2014) 32(7):372–80.
36. Buck PM, Kumar S, Singh SK, Roberts CJ. Influence of colloidal interactions on protein aggregation and viscosity in concentrated antibody solutions. Biotechnol Bioeng. (2013) 110(6):1763–73.
37. Lingg N, Zhang P, Song Z, Bardor M. The sweet tooth of biopharmaceuticals: importance of glycosylation in therapeutic proteins. Biotechnol J. (2012) 7(12):1462–72.
38. Yadav S, Shire SJ, Kalonia DS. Viscosity analysis of high-concentration protein solutions. Mol Pharm. (2010) 7(6):1703–19.
39. Sharma VK, Kalonia DS. Viscosity behavior of high-concentration antibody solutions. J Pharm Sci. (2015) 104(3):1047–56.
40. Esfandiary R, Parupudi A, Sathish HA. Effects of arginine and histidine on viscosity reduction in concentrated monoclonal antibody solutions. Mol Pharm. (2016) 13(10):3658–69.
41. Shukla AA, Trout BL. Interaction of arginine with proteins and the mechanism of viscosity reduction in concentrated antibody solutions. Biophys J. (2011) 101(5):1314–23.
42. Arakawa T, Timasheff SN. The stabilization of proteins by osmolytes. Biochemistry. (1985) 24(25):6756–62.
43. Minton AP. Influence of macromolecular crowding upon the stability and state of association of proteins. Biophys J. (2005) 88:971–85.
44. Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. (2001) 26:597–604.
46. Schuck P. Sedimentation velocity analytical ultracentrifugation: discrete species and size-distribution analysis. Biophys J. (2000) 78:1606–19.
47. Honarvar S, Müller SJ. Microfluidic approaches for high-throughput protein rheology. Lab Chip. (2018) 18:1353–65.
48. Jain T, Sun T, Durand S et al. Biologics developability: the role of computational tools and multi-modal deep learning. Front Pharmacol. (2021) 12:732205.
49. Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP et al. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci U.S.A. (2019) 116:4025–30.
50. Yang Y, Xu T, Yu X et al. Understanding the high viscosity of concentrated monoclonal antibody solutions: structural and interactional insights. Nat Commun. (2022) 13:4089.
51. Kamerzell TJ, Esfandiary R, Joshi SB, Middaugh CR, Volkin DB. Protein-excipient interactions: mechanisms and biophysical characterization applied to protein formulation development. Adv Drug Deliv Rev. (2011) 63:1118–59.
52. Wang W, Singh S, Zeng D, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. (2007) 96:1–26.
53. Roberts CJ. Protein aggregation and viscosity in therapeutic antibody formulations: mechanisms and modeling approaches. Trends Biotechnol. (2014) 32:372–80.
54. Li Y, Ogunnaike BA, Roberts CJ. Multi-scale modeling of protein aggregation kinetics and viscosity behavior in concentrated antibody solutions. Biotechnol Prog. (2010) 26:164–72.
55. Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability using molecular modeling. Proc Natl Acad Sci U.S.A. (2009) 106:11937–42.
56. Bittner B, Richter W, Schmidt J. Subcutaneous administration of biotherapeutics: formulation, device, and delivery considerations. Adv Drug Deliv Rev. (2018) 139:19–33.
57. Roberts CJ. Therapeutic protein aggregation and viscosity: mechanisms and control strategies. Trends Biotechnol. (2014) 32:372–80.
58. Arakawa T, Tsumoto K. The effects of arginine on protein stability and aggregation. Int J Biol Macromol. (2018) 120:1206–14.
59. Wang W, Singh SK, Li N et al. Stabilization of therapeutic proteins by sugars and polyols. Adv Drug Deliv Rev. (2016) 93:89–104.
60. Maa YF, Nguyen PA. Protein stabilization by polysorbates in biopharmaceutical formulations. J Pharm Sci. (2015) 104:2899–914.
61. Byrne N, Angell CA. Protein stabilization by ionic liquids: mechanisms and applications. Chem Commun. (2009):1046–8.
62. Zhao H. Protein stabilization and enzyme activation in ionic liquids: specific ion effects. Chem Soc Rev. (2010) 39:3480–98.
63. Viola M, Sequeira J, Seica R, Veiga F, Serra J, Santos AC. Subcutaneous delivery of monoclonal antibodies: challenges and opportunities. J Control Release. (2018) 286:301–14.
64. Singh SK, Nema S, Wang W. Antibody structure, stability, and formulation. J Pharm Sci. (2007) 96:1–26.
65. Shukla AA, Thömmes J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. (2010) 28:253–61.
66. Frost GI. Recombinant human hyaluronidase (rHuPH20): enabling subcutaneous drug delivery. Drug Deliv. (2007) 14:375–81.
67. Bittner B, Richter W, Hourcade-Potelleret F, Hertel S. Subcutaneous administration of biotherapeutics: formulation and device considerations. Adv Drug Deliv Rev. (2018) 139:19–33.
68. Bittner B, Schmidt J, Tillmann C. Subcutaneous delivery of biotherapeutics: current experience and future perspectives. Adv Drug Deliv Rev. (2021) 172:91–107.
69. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. (2017) 6:576–88.
70. Richter WF, Jacobsen B. Subcutaneous absorption of biotherapeutics: knowns and unknowns. Drug Metab Dispos. (2014) 42:1881–9.
71. Shah DK, Betts AM. Toward a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. (2013) 40:67–86.
72. Mason BD, Zhang L, Remmele RL Jr., Zhang J. Opalescence of an IgG2 monoclonal antibody solution as it relates to liquid–liquid phase separation. J Pharm Sci. (2011) 100(11):4587–96.
73. Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm. (2005) 289:1–30.
74. Wang W, Roberts CJ. Protein aggregation – mechanisms, detection, and control. Int J Pharm. (2018) 550:251–68.
75. Wakankar AA, Borchardt RT. Formulation considerations for proteins susceptible to asparagine deamidation and aspartate isomerization. J Pharm Sci. (2006) 95(11):2321–36.
76. Li S, Schöneich C, Borchardt RT. Chemical instability of protein pharmaceuticals: mechanisms of oxidation and strategies for stabilization. Biotechnol Bioeng. (1995) 48:490–500.
77. Jones LS, Kaufmann A, Middaugh CR. Silicone oil induced aggregation of proteins. J Pharm Sci. (2005) 94(4):918–27.
78. Bee JS, Stevenson JL, Mehta B et al. Tungsten-induced protein aggregation in pharmaceutical formulations. J Pharm Sci. (2011) 100(10):4158–70.
79. Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJA, Russell Middaugh C, Winter G et al. Overlooking subvisible particles in therapeutic protein products: gaps in analytical methods and regulatory considerations. J Pharm Sci. (2009) 98(4):1201–5.
80. Yu LX. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. (2008) 25:781–91.
81. Rathore AS, Winkle H. Quality by design for biopharmaceuticals. Nat Biotechnol. (2009) 27:26–34.
82. Lionberger RA, Lee SL, Lee L, Raw A, Yu LX. Quality by design: concepts for ANDA submissions. AAPS J. (2008) 10:268–76.
83. Rathore AS, Kapoor G. Implementation of quality by design for biologics. Biotechnol Prog. (2017) 33:1489–98.
84. Beg S, Hasnain MS, Rahman M, Swain S. Quality by Design (QbD) approach in pharmaceutical development. Int J Pharm Investig. (2015) 5:1–9.
85. Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK et al. Understanding pharmaceutical quality by design. AAPS J. (2014) 16:771–83.
86. Usmani SZ, Nahi H, Mateos MV et al. Subcutaneous daratumumab versus intravenous daratumumab in patients with multiple myeloma (COLUMBA study). Lancet Haematol. (2020) 7:e370–80.
87. Cohen SB, Tanaka Y, Mariette X et al. Long-term safety of adalimumab citrate-free formulations in rheumatoid arthritis. Ann Rheum Dis. (2018) 77:180–7.
88. Burmester GR, Rigby WF, van Vollenhoven RF, Kay J, Rubbert-Roth A, Kelman A et al. Tocilizumab in early progressive rheumatoid arthritis: function and safety of subcutaneous administration. Ann Rheum Dis. (2014) 73:69–74.
89. Blauvelt A, Simpson EL, Tyring SK et al. Dupilumab therapy for atopic dermatitis: clinical efficacy and safety. N Engl J Med. (2017) 376:2335–48.
90. Reich K, Papp KA, Blauvelt A et al. Secukinumab efficacy and safety in plaque psoriasis. Lancet. (2015) 386:1137–46.
© The Author(s). 2026 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.